
Obwohl die DNA und ihre Doppelhelix zu den bekanntesten Molekülen unserer Zeit gehören, ist unser Wissen darüber, wie Zellen kontrollieren, welche Gene sie exprimieren wollen, noch recht begrenzt. Um etwa ein Enzym herzustellen, muss die Information, die in unsere DNA über dieses Enzym eingeschrieben ist, zunächst transkribiert und in die Sprache anderer Moleküle „übersetzt“ werden. Um diesen hochkomplexen Prozess zu starten, binden spezielle regulatorische Proteine, genannt Transkriptionsfaktoren (TF), an bestimmte DNA-Regionen. Auf diese Weise können sie die Expression eines Gens an- und abschalten. Die große Frage ist: Wie können Transkriptionsfaktoren die richtige Stelle auf der DNA finden, um die Genexpression zu regulieren?
Einfaches Modell – große Wirkung
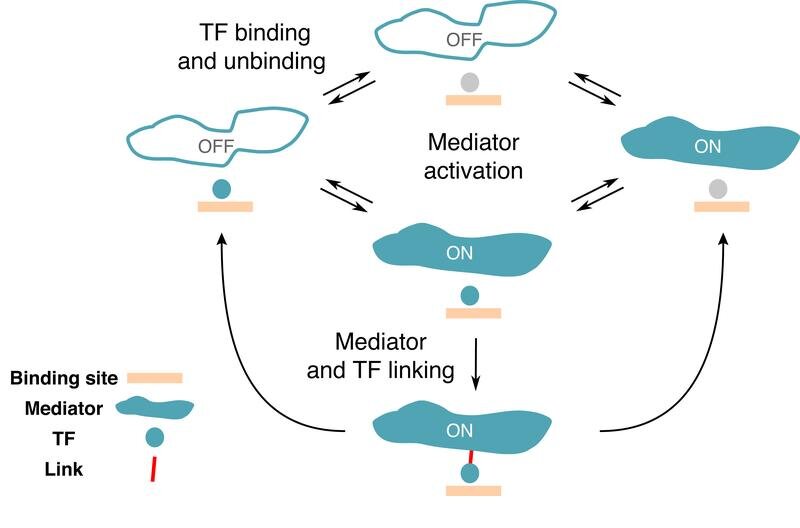
Bereits heute sind biophysikalische Modelle in der Lage, die Genexpression anhand des Zusammenspiels von TFs und DNA-Regulationsregionen vorherzusagen. Allerdings nur bei Prokaryonten, einfachen zelluläre Organismen ohne Zellkern, wie etwa Bakterien. Bei Prokaryonten sind die TF-Bindungsstellen auf der DNA vergleichsweise lang und spezifisch, was es den TFs erleichtert, ihr Ziel zu finden. Bei höheren Organismen, den sogenannten Eukaryonten, deren Zellen einen Kern haben, ist die mathematische Beschreibung des Prozesses der Genregulation deutlich schwieriger. Ein Forscherteam am Institute of Science and Technology Austria (IST Austria) hat nun einen Weg gefunden, um zu beschreiben, wie die Interaktion zwischen den verschiedenen regulatorischen Molekülen in Eukaryonten aussehen könnte.
In einer neuen Studie, die in PNAS veröffentlicht wurde, haben Rok Grah in Zusammenarbeit mit IST-Professor Gašper Tkačik und Benjamin Zoller von der Universität Princeton eine minimale Erweiterung eines klassischen Gleichgewichtsmodells vorgeschlagen, das auf den Wechsel zwischen dem aktiven und inaktiven Zustand eines Gens angewendet werden kann. Zu diesem Zweck wählten sie eine Reihe von Merkmalen oder „regulatorischen Phänotypen" aus, mit denen das gewünschte Modell umgehen können sollte. „Wir wollten ein Gen mit hoher Spezifität, was bedeutet, dass das Gen nur durch die richtigen TFs aktiviert wird", sagt der ehemalige PhD-Student am IST Austria Rok Grah. Ein weiteres Merkmal, das in das Modell einbezogen wurde, war die Verweildauer der TFs auf einer spezifischen sowie einer zufälligen DNA-Region. „Wir konnten zeigen, dass es eine Klasse von einfachen Modellen gibt, die bei all diesen Phänotypen gut funktionieren, was bisher noch nicht gelungen ist", erklärt Benjamin Zoller. Auch wenn die vorgeschlagene Erweiterung des klassischen mathematischen Modells sehr klein ist, zeigt sich so eine Fülle qualitativ neuer Verhaltensweisen, die mit den aktuellen experimentellen Einschränkungen übereinstimmen.
Verrauschte Gene
Auf Basis der so gewonnenen Daten argumentieren die Forscher, dass einzelne TFs nur begrenzt zwischen spezifischen und zufälligen Stellen auf der DNA unterscheiden können. Jeder TF-Typ bindet bevorzugt an bestimmte regulatorische DNA-Sequenzen. Sie binden aber auch an andere, nicht verwandte Ziele. „Unsere Hauptmotivation war es, ein Modell zu finden, das beschreibt, wie die regulatorischen Elemente auf der DNA nicht durch „fremde“ Transkriptionsfaktoren aktiviert werden", so Benjamin Zoller. Ihre Ergebnisse legen nahe, dass eine hohe Spezifität der Genexpression eine gemeinsame Leistung der regulatorischen Moleküle während des „Korrekturlesens“ sein muss.
Wenn ein Gen aktiv ist, schwankt zudem die Anzahl der von ihm produzierten Proteine, was von den Wissenschaftlern als Genexpressionsrauschen bezeichnet wird. „Was mich überrascht hat, war der Zusammenhang zwischen Rauschen und Spezifität. Wenn man eine hohe Spezifität haben will, scheint es so zu sein, dass dies tendenziell zu mehr Rauschen führt, was faszinierend ist", sagt Benjamin Zoller. Ein starkes Rauschen wird oft als schädlich für Zellen angesehen, doch Gene in Eukaryonten seien ziemlich verrauscht. "Bis jetzt wissen wir nicht wirklich, warum sich diese ganze Transkriptionsmaschinerie so entwickelt hat. Vielleicht ist eine Erklärung dafür, dass hohes Rauschen unvermeidlich ist, wenn man eine hohe Spezifität erreichen will. In unserem Modell scheint es keinen Ausweg zu geben. Hohe Spezifität bedeutet immer hohes Rauschen, und es ist möglich, dass Zellen Mechanismen entwickelt haben, um das Rauschen später im Genexpressionsprozess zu verringern", fügt Datenwissenschaftler Rok Grah hinzu. In einem nächsten Schritt wollen die Forscher das neue Modell experimentellen Tests unterziehen. Aufgrund seiner Einfachheit eignet es sich perfekt für die Arbeit mit präzisen Echtzeit-Genexpressionsmessungen, zum Beispiel von DNA-Regulationssequenzen, die bewusst verändert wurden.
IST Austria
Originalpublikation:
Grah R, Zoller B, Tkačik G. 2020. Non-equilibrium models of optimal enhancer function. PNAS.

 in ihr Dorf im kolumbianischen Amazonas-Gebiet nahe des Apaporis-Flusses.")
")