
Ein internationales Forscherteam unter Federführung der Universität Bonn hat die Ursache einer seltenen, schweren Muskelerkrankung aufgeklärt. Eine einzelne spontan auftretende Mutation führt demnach dazu, dass die Muskelzellen defekte Proteine nicht mehr korrekt abbauen können. In der Folge gehen die Zellen zugrunde. Das Leiden führt bei Kindern zu einer schweren Herzschwäche, begleitet von Schäden der Skelett- und Atem-Muskulatur. Die Betroffenen werden selten älter als 20 Jahre. Die Studie zeigt auch experimentelle Ansätze für eine mögliche Therapie auf. Ob sich diese Hoffnung erfüllt, wird sich allerdings erst in einigen Jahren zeigen.
Wer am Fahrrad schon einmal einen Speichenbruch hatte oder mit dem Auto liegengeblieben ist, der weiß: mechanische Belastungen führen früher oder später zu Schäden, die repariert werden müssen. Das trifft auch auf die menschliche Muskulatur zu. „Bei jeder Bewegung werden Strukturproteine geschädigt und müssen ersetzt werden“, erklärt der Privatdozent Dr. Michael Hesse vom Institut für Physiologie der Universität Bonn, der die Studie zusammen mit seinem Kollegen Prof. Dr. Bernd Fleischmann geleitet hat.
Normalerweise werden die defekten Moleküle in der Zelle zerkleinert und ihre Bestandteile dann recycelt. Eine wichtige Rolle bei diesem komplexen Prozess spielt ein Protein namens BAG3. Wie wichtig, zeigen die Ergebnisse der neuen Studie: Die Forschenden konnten darin nachweisen, dass eine einzige Veränderung in der genetischen Bauanleitung von BAG3 zu einer tödlichen Erkrankung führt.
„Die Mutation führt dazu, dass BAG3 zusammen mit Partner-Proteinen unlösliche Komplexe bildet, die immer größer werden“, sagt Hesse. Dadurch kommen die Reparaturprozesse zum Erliegen – die Muskulatur wird immer weniger leistungsfähig. Zudem entsteht mit der Zeit eine toxische Protein-Anreicherung, die schließlich zum Tod der Muskelzelle führt. „Die Folgen werden meist zuerst am Herz sichtbar“, sagt Hesse. „Dort wird Muskel sukzessive durch Narbengewebe ersetzt. Dadurch sinkt die Elastizität des Herzens, bis es kaum noch Blut pumpen kann.“
Bei den Betroffenen ist daher meist schon im Kindesalter eine Herztransplantation erforderlich. Dennoch hilft diese Maßnahme ebenfalls nur temporär, da die Krankheit auch die Skelett- und Atemmuskulatur erfasst. Oft sterben die Erkrankten daher schon in jungen Jahren.
Sehr seltenes Leiden, daher wenig Forschung
Die tödliche Mutation kann spontan bei der Entwicklung des Embryos entstehen. Glücklicherweise tritt das sehr selten auf: Weltweit gibt es vermutlich nur wenige hundert Kinder, die betroffen sind. Aufgrund ihrer Seltenheit wurde die Erkrankung bislang aber kaum erforscht. „Unsere Studie bringt uns nun ein großes Stück weiter“, betont Bernd Fleischmann.
Denn den Forschenden ist es gelungen, die Erkrankung erstmals in Mäusen nachzustellen und mit Hilfe des neuen Tiermodells ihre Ursachen aufzuklären. Dadurch lässt sie sich besser erforschen als bislang – auch im Hinblick auf mögliche Therapien. Denn eventuell lässt sich die Auswirkung der Mutation zumindest mindern. Menschen haben von jeder Erbanlage zwei Versionen, von denen die eine von der Mutter und die andere vom Vater stammt. Selbst wenn bei der Entwicklung des Embryos eine BAG3-Version mutiert, gibt es also immer noch ein zweites Gen, das intakt ist.
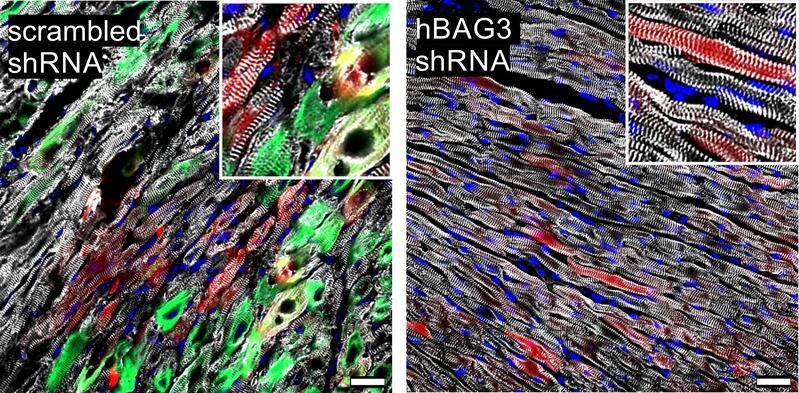
Leider verklumpt jedoch das defekte BAG3 auch mit seinen intakten Geschwistern. Die Mutation in einem der Gene reicht also, um den Abbau der defekten Muskelproteine zum Stillstand zu bringen. Könnte man die mutierte Version aus dem Verkehr ziehen, müsste die Reparatur aber wieder funktionieren. Außerdem ließe sich so die massive Anhäufung von Proteinen in der Zelle verhindern, die schließlich zu ihrem Tod führt.
Tatsächlich gibt es Methoden, einzelne Gene in ihrer Aktivität gezielt zu hemmen. „Wir haben eine davon genutzt, um die kranken Mäuse zu behandeln“, erklärt Kathrin Graf-Riesen vom Institut für Physiologie, die zusammen mit Dr. Kenichi Kimura und ihrer Kollegin Dr. Astrid Ooms für einen großen Teil der Experimente verantwortlich war. Die so therapierten Tiere zeigten dann deutlich weniger Symptome. Ob sich dieser Ansatz auf den Menschen übertragen lässt, ist allerdings noch Gegenstand weiterer Forschung.
Universität Bonn
Originalpublikation:
Kenichi Kimura et al.: Overexpression of human BAG3P209L in mice causes restrictive cardiomyopathy. Nature Communications, https://doi.org/10.1038/s41467-021-23858-7

 in ihr Dorf im kolumbianischen Amazonas-Gebiet nahe des Apaporis-Flusses.")
")